- 5-alpha reductase deficiency
- Androgen Insensitivity Syndrome (AIS)
- Aphallia
- Clitoromegaly (large clitoris)
- Congenital Adrenal Hyperplasia (CAH)
- gonadal dysgenesis (partial & complete)
- hypospadias
- Klinefelter Syndrome
- micropenis
- mosaicism involving “sex” chromosomes
- MRKH (Mullerian agenesis; vaginal agenesis; congenital absence of vagina)
- ovo-testes (formerly called “true hermaphroditism”)
- Partial Androgen Insensitivity Syndrome (PAIS)
- Progestin Induced Virilization
- Swyer Syndrome
- Turner Syndrome
Klinefelter sendromu
47, XXY sendromu olarak da bilinir. Hhücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.
Hastalığı, 1942’de Dr. Harry Klinefelter ilk olarak tanımlamıştır, doktorun adıyla anılan doğuştan gelen bir hastalıktır.
Bu durumdaki kişiler genellikle erkek birey olarak görülür. Uzun kol ve bacakları, kadınımsı kalça çıkıntıları ilk olarak göze çarpan özellikleridir. Testisleri küçük, kadınımsı göğüs (jinekomasti) ve kas gelişimleri vardır. Sesleri erkeklere nazaran daha incedir. Hipogonadizm görülebilir. Sakal ve bıyık gelişimleri çok az, vücut kıllanmaları kadınımsı görünümdedir. Spermatogenez görülmez ve kısır bireylerdir.
Canlı erkek doğan bireylerin 500 ya da 1000’inde 1 oranla görülür.
***
48, XXYY sendromu; yaklaşık 17,000 canlı doğumda bir karşılaşılan Klinefelter sendromunun özelliklerini taşır.
Mozaik 47,XXY/46,XY Klinefelter sendromu; şeklinde mozaik olarak da görülebilir. Bu kişilerde semptomlar 47,XXY karyotipli hücrelerin yoğunluğuna bağlı olarak değişir.
***
Swyer Sendromu
Swyer Sendromu, XY cinsel salgı bezlerinin embriyo evresindeki “anormal” organ gelişimi olarak da bilinir. Kişide işlevli cinsel salgı bezleri yoktur. Bunların yerine, testis veya yumurtalıkta az gelişmiş/işlevsiz cinsel salgı bezi yer alır. Swyer ile doğmuş bir çocuk kadına benzer. (doktor tarafından reçete edilerek) hormon tedavisi olmazsa birçok ikincil cinsiyet özellikleri gelişmez çünkü işlevsiz cinsel salgı bezleri ergenliği başlatan cinsiyet hormonları (hem androjen hem östrojen) üretemez. Erkeklerde testis yapısı, kadınlarda da aylık adet döngüsü görülmez, hatta kadınlarda vajina yapısında olgunlaşmamış testisler görülebilir.
***
Androjene Duyarsızlık Sendromu
Androjene Duyarsızlık Sendromu, ADS, bir genetik durumdur, ( kendiliğinden oluşan mutasyonlar dışında) kalıtsaldır, yaklaşık olarak 20.000 kişiden bir kişide görülür. Testiküler feminizasyon veya androjen insensitivitesi olarak da bilinir. Androjen reseptör mekanizması bozukluğu nedeniyle vücut hücreleri androjene tepki verememektedir ( “Erkek hormonları” tabiri, bu hormonlar hem kadında hem de erkekte sürmekte ve aktif olduğundan yersizdir.
XY genotipli ve testisi olan birey normal dişi görünümündedir. Vajina vardır fakat kör bir kese halindedir, rahim ve fallop tüpleri oluşabilir, ergenlikte meme gelişimi gözlenir; fakat regl olmaz, kısırdırlar, pubik kıllar azdır ya da yoktur. Testisler genelde abdomende ve embriyolojik dönemde aşağı inerken kullandıkları geçiş yolu üzerinde, inguinal kanalda yerleşmiştir.
Bazı kişilerde de kısmı androjene duyarsızlık vardır. Klitoris ve dış genital organlar daha erkeksi gelişmiştir, vajina yine kör olarak sonlanır, rahim yoktur.
Testisler ileri yaşlarda kötü huylu tümöre dönüşme olasılıkları vardır. Tespit edilse de kişi ameliyat kararı alabilecek yaşa geldiğinde tavsiye edilmesi daha yerinde olur. Ergenlik öncesi testis kanseri olma ihtimali çok düşüktür. Ölümcül risk faktörleri pek fazla değildir, ömür boyu takip edilmesi gereken sorunlar ortaya çıkmaz.
ADS sahip bebekler veya genç kızlara, vajina derinliğini arttırmak için vajinoplasti ameliyatı yapılması çok yaygındır. Böylece gerekli derinliğe sahip olunca uygun büyüklükteki bir penisi içlerine alabilirler. Vajinoplasti ameliyatları başarısız olmaları sebebiyle sorunludur. Bu tür ameliyatlar, ergenlik çağındaki bir kıza tavsiye edilmeli ama dayatılmamalıdır. Karar vermeden önce ADS’li bir kadınla cinsel tecrübeleri ve ameliyat hakkında bilgi alması daha yerinde olur. Her ADS’li kadın ameliyat olmayı seçmemektedir.
Doktorlar ve ebeveynler ADS kızlara durumları hakkında dürüst olmak konusunda çok isteksizler. Bu gizlilik ve yaftalama kendini farklı hissetme duygusunu gereksiz yere arttırmaktadır.
Çünkü ADS X kromozomundaki genetik bir bozukluk olduğu için ailede kuşaktan kuşağa geçer.
***
Doğuştan Böbrek Üstü Bezi Büyümesi
Doğuştan Böbrek Üstü Bezi Büyümesi, XX kromozomlarıyla doğmuş kişilerde görülen en yaygın interseks durumudur. XY kromozomda görülmediği için yaygınlığı 20.000 ile 36.000 de birdir. İnterseks durumları içinde, bebekken tıbbi müdahale gerektiren tek durumdur.
Böbrek üstü bezlerinde (birçok hormon üretir ve onları kan dolaşımına bırakır) kortizon üreten gen “tarifinin” bozulması sonucu Doğuştan Böbrek Üstü Bezi Büyümesi oluşur. Tarif bozulduğu için, böbrek üstü bezleri kortizon üretmeye çalışırken kana karışan çok yüksek oranda hormon üretir. Bu, XX embriyolarının daha büyük klitorislerinin olması ve hatta penise benzeyen klitorislerinin olması ya da testis torbasına benzer vajina dudakları olması demektir. Kana karışma metabolizma kaynaklı olduğu için erkeksileşme etkileri doğumdan sonra da devam eder. Yani Doğuştan Böbrek Üstü Bezi Büyümesi sık vücut kılı, açılan alın bölgesi, kalın ses, belirgin kaslar gibi kişinin erkeksi özellikler geliştirmesine neden olur.
***
Aphallia
Penisi olmadan doğmak demektir. Bunun dışında beden erkek anatomisindedir.
***
Clitoromegaly (büyük klitoris)
Beklenenden daha büyük klitoris demektir. Bir tanıdan çok tariftir. Büyük klitorisin en yaygın nedeni Doğuştan Böbrek Üstü Bezi Büyümesidir.
***
Hypospadias
Halk arasında Peygamber Sünneti olarak da bilinen Hypospadias, idrar deliğinin normalden daha aşağıda olmasıdır. Tüm erkek çocukların % 1-2’sinde gözlenir. % 10-15 ailevi yatkınlık gösterir.
***
Mikro-penis
Penisin normalden daha küçük olması durumuna verilen addır. Mikro-penis klinik tanısı; 46 XY kromozoma sahip (eril kromozom), testisler inmiş veya inmemiş, penis ucunda urethral meatus (idrar yolu ) ve 3-7 santimde veya altında penis boyudur.
Tedavilerinin farklı olmasından dolayı mikropenisin 2 tipi vardır. 3-7 santim penislerde, penisi vücuda bağlayan bağların kesilerek, 2 santim uzama sağlanıyor. 2-3 santim olan ikinci tip mikropenislerde ise ön koldan alınan doku penis şekline getiriliyor. Tedavi isteğe bağlıdır.
***
Mullerian agenesis; vaginal agenesis; doğuştan vajinasız olmak
Yumurtalıklar var ama ya rahim yok ya da yarı gelişmiş, çok az bir azınlıkta bunlara böbrek ve omurilik anormallikleri eşlik edebilir.
***
Ovo-testes (eskiden “gerçek hermafroditizm” adlandırılırdı)
Bir bölümü ovaryum bir bölümü testis dokusu içeren iç genital yapı. Tek bir yumurtalığın veya her ikisinin, tek bir testisin veya her ikisinin de içinde olabilir.
***
Cinsiyet kromozomlarında mozaisizm
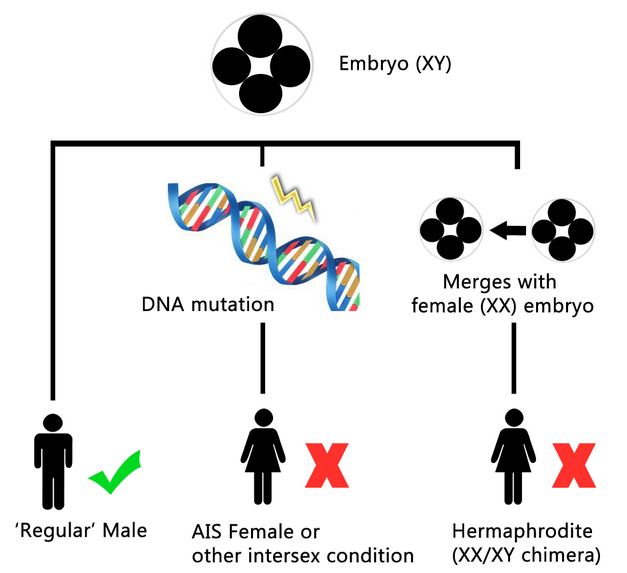
Öncelikle biraz arkaplanına bakalım: Bir karyotip bir hücredeki kromozomların resmidir. Karyotip kişinin ne tür kromozomlar taşıdığını bulmak için kullanılır. Bu, kişiden kan ya da doku örneği alınıp örnek boyanarak mikroskopta görüntülenerek gerçekleştirilir. Görüntü daha sonra kesilir ve yeniden düzenlenir ve kromozomlar birbirine denk çiftler şeklinde dizilir. Çıkan sonuç kişinin kromozomlarının sayısı ve tipi olarak açıklanır, örneğin 45,X (birey 22 çift eş kromozom ve bir X kromozomuna sahip anlamına gelir, bu durum Turnet Sendromu olarak adlandırılır); 46;XX (birey 22 çift eş kromozom ve 2 X kromozoma sahip anlamına gelir); 46,XY (birey 22 çift eş kromozoma ve 1 X, 1 Y kromozomuna sahip anlamına gelir); 47, XXY.
Bir birey hücrelerinin bazılarında belli bir karyotip, bazılarında başka bir karyotip taşıdığında bu durum “mozaik karyotip” olarak sınıflandırılır. Örneğin, kişinin 45,X/46,XX karyotip olduğu söylendiğinde bu kişinin bazı hücrelerinde 46,X karyotip, bazı hücrelerinde 46,XX karyotip taşıdığı anlamına gelir. Mozaisizm embriyonun erken dönemlerinde hücrelerin yanlış bölünmesinden kaynaklanır.
Örneğin Mozaik Turner sendromlu bir kişi XO karyotipli bazı hücreler taşıyabilir (tipik Turner sendrom karyotipi) bunun yanı sıra XX karyotipinde (tipik dişi karyotipi) bazı hücreler de taşıyabilir, bazı durumlarda diğer karyotip XY de olabilir. Mozaisizm ayrıca daha yumuşak biçimlerde 46/47 XY/XXy mozaik olarak adlandırılan Klinefelter Sendromunda da görülür. Bu durumda, XY hücreler 46 kromozom taşıyacaktır (tipik kromozom sayısı) ve XXY hücreler 47 kromozom taşıyacaktır.
***
Turner Sendromu
Bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkan çeşitli semptomların tümüne verilen addır.
Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir. Turner sendromlu bireylerde doğuştan böbrek rahatsızlıkları, kalp anomalileri, kistik higroma en çok görülen hastalıklardır.
Kısa ve perdeli boyun
Kısa boy
Limfodema el ve ayaklarda şişkinlik
Göğüs kafesi farklılığı ve memeucu genişliği
Düşük saç bitişi
Düşük kulak çizgisi
Kısırlık
Amenore (adet görmeme)
Diğer belirtiler, dışa kıvrılan dirsek (kübitus valgus), perdeli boyun, yumuşak ve yukarı dönen tırnaklar, Simian çizgisi ve düşük gözkapaklarıdır. Turner sendromu bulguları kişiden kişiye farklılık gösterebilir.
Çeviri: Aligül Arıkan
